Chemistry AI for Autonomous Discovery
Molecule.one's Maria™ unites AI, Lab, and Data to power near-autonomous research for drug discovery & manufacturing. Commercially proven.

Chan-Lam Synthesis Discovery OA1-M1-003

In a first for organic chemistry, Molecule.one & OpenAI have successfully put AI to work on an open-ended scientific problem. GPT-5.4 & Maria AI picked the research area, generated proposals, rated them, and ran the experiments in the Maria Lab.
OpenAI post | Preprint
The Maria™ AI platform

Maria AI
Frontier chemistry models combined in an agentic framework to solve chemical problems and discover new chemistry

Maria Lab
The revolutionary, micro-liter scale HTE lab directed by Maria AI and tailored for her workflows

Maria Data
The industry leading, novel data set generated at Maria AI's direction in her purpose-built lab
Press Highlights

A near-autonomous AI chemist improves a challenging reaction in medicinal chemistry
June 17, 2026

Grace and Molecule.one to Transform Peptide Building Block Synthesis with AI and Automation
December 8, 2025

Standard Industries Announces Molecule.one as Winner of $1 Million AI Challenge
June 12, 2025

CAS and Molecule.one Announce a Strategic Collaboration to Accelerate Drug Discovery
August 11, 2023
Papers
JCIM, 2021
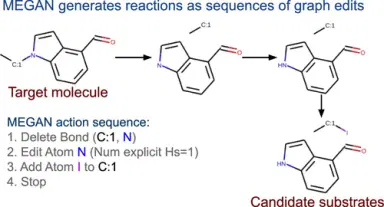
Molecule Edit Graph Attention Network: Modeling Chemical Reactions as Sequences of Graph Edits
2021
EFMC-ASMC 2025
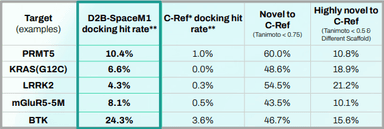
10× Higher Hit Rates At 3–10× Lower Cost: Accelerating Ligand Discovery With ML-Guided Synthesis & Screening
2025
NeurIPS Workshop AI4Science, 2025
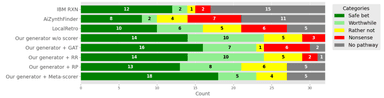
Trustworthy Retrosynthesis: Mitigating Hallucinations with Reaction Plausibility Filtering and Retrieval-Augmented Scoring
2025
NeurIPS Workshop AI4Science, 2025
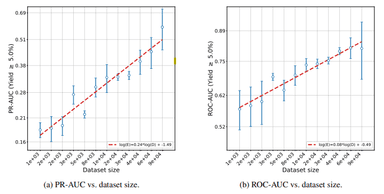
Scaling High-Throughput Experimentation Unlocks Robust Reaction-Outcome Prediction
2025
Journal of Cheminformatics, 2024
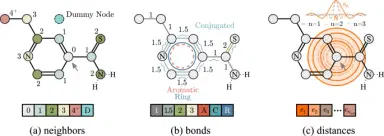
Relative molecule self-attention transformer
2024
Case studies

Engineering Serendipity: Rapid Single-Iteration Hit Discovery using Plate Analogs in μSpaceM1
Case Study

First Application of AI-Driven Small Molecule Synthesis in a Non-Pharmaceutical Industry
Case Study

Accelerating Hit-to-Lead Campaigns with molecule.one
Case Study

Rapid, Single-Iteration Hit Identification for CLK1 using Direct-to-Biology
Case Study
Posts

The future of autonomous science may be both human-like and machine-like
July 2, 2026

Trustworthy Retrosynthesis: How a Culture of Chemist-AI Collaboration Won $1M and Led to a Strategic Partnership with W.R. Grace
December 11, 2025

How Did a Startup End Up Running World's Largest Microliter-Scale HTE Reaction Campaign?
October 31, 2025